Ich muss was zugeben. Ab jetzt bin ich ein evolutionärer Kreationist. Ich habe keine Wahl. Der Beweis für biologische Evolution ist so überwältigend…
…April! April! Nur ein Scherz.
Ich bin immer noch ein Old-Earth Kreationist. Obwohl das Evolutionsparadigma die übergeordneten Rahmenbedingungen in der Biologie setzt, bleibe ich skeptisch gegenüber Facetten davon. Ich bin überzeugter als je zuvor, dass ein Ansatz, der auf ein Schöpfungsmodell rührt, die beste Perspektive fürs Verstehen der Entstehung, Geschichte und des Designs vom Leben bietet. Damit sage ich nicht, dass es keinen Beweis für gemeinsame Abstammung; den gibt’s.Aber auch mit diesem Beweis halte ich Old-Earth Kreationismus aus drei Gründen für überlegen.
Erstens: Ein Schöpfungsmodell kann sehr einfach den Beweis für gemeinsame Abstammung im Rahmen des Designs unterbringen.
Zweitens tut sich das Paradigma der Evolution schwer, adäquate Erklärungen für die wichtigen Übergänge in der Geschichte des Lebens zu finden.
Drittens ist der Eindruck des Designs überwältigend—und wird mit jedem Tag noch überwältigender.
Das ist kein Witz.
Das menschliche Genom ist ein Beispiel. Wenn es darum geht, seine Struktur und Funktion zu verstehen, stecken wir noch in den Windeln. Als unser Wissen und unsere Einsicht wachsen, wird es zunehmend klar, dass die strukturellen und funktionellen Eigenschaften des menschlichen Genoms (und die Genome anderer Organismen) mehr Eleganz und Ausgefeiltheit aufweisen, als die meisten Naturwissenschaftler sich hätten vorstellen können—mindestens, diejenigen die innerhalb vom Rahmen der Evolution arbeiten. Demgegenüber wird die Eleganz und Ausgefeiltheit des Genoms von Kreationisten und Verfechter des Intelligent Designs erwartet. Einfach gesagt, je mehr wir über das menschliche Genom lernen, desto mehr scheint es das Produkt eines Geistes zu sein.
Die Tatsache ist, der Fortschritt in Genomik im letzten Jahrzehnt hat Naturwissenschaftler gezwungen, ihre Ansichten der Genombiologie zu ändern. Als das menschliche Genom in 2000 zuerst sequenziert wurde, dachten die Biologen, dass die Mehrheit der sequenzierten Elemente nicht funktionell sein und aus nutzloser DNS bestehen würden. Jetzt erkennen Biologen, dass fast jede Klasse dieser so-genannten Junk-DNS-Sequenzen Schlüsselrollen und Funktionen hat.
Wenn die meisten DNA Sequenzelemente im menschlichen Genom wirklich “Junk DNA” wären, würde ich damit übereinstimmen, dass man sie als Überreste der Evolutionsgeschichte betrachten soll, besonders, weil diese Junk- DNS Sequenzen in den entsprechenden Stellen in den Genomen der Menschen und Menschenaffen erscheinen. Aus diesen Gründen haben Biologen traditionell diese gemeinsamen Sequenzen für den überzeugendsten Beweis für die gemeinsame Abstammung interpretiert.
Aber ich denke, jetzt, da wir gelernt haben, dass diese Sequenzen funktionell sind, dass es völlig vernünftig ist, sie als das Handwerk eines Schöpfers zu betrachten, und zu erkennen, dass sie absichtlich entworfen wurden, der Biologie des Genoms beizutragen. In diesem Rahmen widerspiegeln diese gemeinsamen DNS-Sequenzen in den Genomen der Menschen und Menschenaffen gemeinsames Design, nicht gemeinsame Abstammung.
Dennoch lehnen viele Biologen die Interpretation des gemeinsamen Designs ab, weil sie weiterhin lieber dem evolutionären Modell vertrauen. Ihr Vertrauen widerspiegelt eine Verpflichtung dem methodologischen Naturalismus gegenüber, aber es gibt einen anderen Grund für Ihr Vertrauen. Sie argumentieren, dass das menschliche Genom (und die Genome anderer Organismen) andere architektonischen und operationellen Eigenschaften aufweisen, die bestens durch den evolutionären Rahmen erklärt werden—und, ihrer Ansicht zufolge– dass diese Eigenschaften die Waagschale der Beweisführung zu Gunsten der evolutionären Interpretation kippen.
Aber Forscher machen weitere Entdeckungen über Junk DNS, die den Beweis für gemeinsame Abstammung ausgleichen, unter anderen diese strukturellen und funktionellen Eigenschaften. Neue Einsichten in die Pseudogenbiologie veranschaulichen diesen Trend sehr schön.
Pseudogene
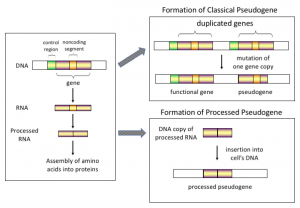
Die meisten Naturwissenschaftler halten Pseudogene für die Überreste einst funktioneller Gene. Dementsprechend haben Biologen drei Kategorien von Pseudogenen bestimmt (unitär, dupliziert und bearbeitet) und drei unterschiedliche Mechanismen vorgeschlagen, die die Entstehung jeder Klasse erklären. Diese Mechanismen erzeugen Unterscheidungsmerkmale, die es den Investigatoren befähigt, gewisse DNS-Sequenzen als Pseudogene zu identifizieren. Aber eine a priori Verpflichtung zum Paradigma der Evolution kann viele Biologen dazu bewegen, übereilig den Schluss zu ziehen, dass diese Pseudogene wegen ihren Sequenzeigenschaften nicht funktionell sind. 1 Siehe Abbildung
Die mechanismen der Pseudogenformation Bildnachweis: Wikipedia
Der alte Spruch lautet: Theorie leitet, Experiment entscheidet. Es häufen sich Versuchsdaten, die zeigen, dass Pseudogene aus allen drei Klassen ihren Nutzen haben.
Einige Forscherteams haben gezeigt, dass die Maschinerie der Zelle die bearbeiteten Pseudogenes transkribieren und diese Transkriptionen wiederum in Proteine übersetzt werden. Duplizierte und unitäre Pseudogene werden auch transkribiert. Mit wenigen Ausnahmen werden diese Transkripte aber nicht in Proteine übersetzt. Die meisten duplizierten und unitären Pseudogentranskripte spielen eine regelnde Rolle, die durch die Hypothese der kompetitiven endogenen RNS beschrieben wird.
Mit anderen Worten: Die Unterstützung für Pseudogenfunktion aus Experiment scheint von der Transkription dieser Sequenzen abzuhängen. Das führt zur Frage: Was geschieht mit Pseudogensequenzen, die sich in Genomen befinden, die nicht transkribiert werden? Eine Anzahl pseudogenischen Sequenzen scheinen inaktiv zu sein. Sie werden nicht transkribiert und haben, man nimmt an, keinen Nutzwert überhaupt.
Viele Naturwissenschaftler halten das für unterstützenden Beweis für die evolutionäre Erklärung der Entstehung der Pseudogene und ziehen diese Erklärung jedem Modell vor, das eine Absicht und Design hinter den Pseudogensequenzen vorschlägt. Warum würde ein Schöpfer mutationsbeschädigte Gene einführen, die keine Funktion haben? Bietet Neofuktionalisation nicht eine bessere Erklärung fürs Vorhandensein von funktionsfähigen Pseudogenen, wobei evolutionäre Mechanismen abgearbeitete Pseudogene übernehmen und die dann als Rohstoff fürs Weiterentwickeln neuer DNS Sequenzelemente in neue Gene benutzen?
Oder vielleicht ist es besser, die Transkriptionen der regulierenden unitären und duplizierenden Pseudogene als die funktionellen Überreste der originellen Gene, deren Transkripte eine Rolle in den regulierenden Netzwerken mit anderen RNS-Transkripten spielten, zu betrachten? Obwohl diese Pseudogene Proteinproduktion nicht mehr lenken, können sie immer noch an die aus RNS-Transkripten bestehenden regulierenden Netzwerken teilnehmen.
Sind untranskribierte Pseudogene wirklich untranskribiert?
Denken wir noch mal dran, dass die Unterstützung der evolutionären Deutung der Pseudogene auf der Inferenz gründet, dass einige Pseudogene nicht transkribiert werden. Was wird aus dieser Unterstützung, wenn diese DNS-Sequenzen doch transkribiert werden und wir einfach bis jetzt ihre Transkripte experimentell weder entdeckt noch identifiziert haben?
Ein Hinweis in diese Richtung erschien in einem Artikel für Nature Reviews. In dem Artikel argumentiert ein Team wissenschaftlicher Mitarbeiter aus Australien, dass Versuche, Pseudogentranskripte experimentell zu entdecken, scheitern, bedeutet nicht, dass keine Transkripion existiert.2 Zum Beispiel, die Transkripte eines Pseudogens, das auf einem niedrigen Niveau transkribiert wird, können unter die experimentelle Nachweisgrenze fallen. So ein Pseudogen würde den Forschern inaktiv scheinen, obwohl es tatsächlich aktiv wäre. Darüber hinaus könnte Pseudogenenexpression gewebespezifisch sein oder nur zu gewissen Punkten im Wachstum-und-Entwicklungsprozess stattfinden. Wenn die Untersuchung diesen Möglichkeiten nicht Rechnung trägt, dann könnte das Scheitern der Versuche, Pseudogenetranskripte aufzuspüren, nur bedeuten, dass das Experimentprotokoll fehlerhaft ist.
Die Ähnlichkeit der DNS-Sequenzen der Pseudogene und ihrer entsprechenden “Schwester” -Genen bringt mit sich eine weitere Komplikation. Es kann schwierig sein, experimentell zwischen einem Pseudogene und seinem “intakten” Schwester-Gen zu unterscheiden. Diese Einschränkung bedeutet, dass, in manchen Fällen, Pseudogenetranskripte fälschlicherweise als die Transkripte des “intakten” Gens identifiziert werden. Dies kann wiederum dazu führen, dass Forscher irrtümlicherweise schlussfolgern, dass das Pseudogen nicht transkribiert wird.
Sind untranskribierte Pseudogene wirklich nicht funktionsfähig?
Diese experimentellen Herausforderungen sind aber real, und es gibt tatsächlich Pseudogene, die nicht transkribiert werden, aber es wäre falsch, zu schlussfolgern, dass sie keine Rolle in der Genregulation spielen. Zum Beispiel, ein großes internationales Forschungsteam hat bewiesen, dass eine Pseudogensequenz zur spezifischen drei-dimensionalen Architektur der Chromosomen beiträgt. Dadurch übt diese Sequenz Einfluss über Genexpression aus, wenn auch indirekt. 3
Ein weiteres Forschungsteam stellte fest, dass ein anderes Pseudogen eine Rolle im Aufrechterhalten chromosomaler Stabilität spielt. In Laboruntersuchungen haben sie entdeckt, dass, wenn man die DNS-Region mit diesem Pseudogen tilgt (was man “Deletion” nennt), das erhöht die Anzahl der chromosomalen Recombinationsereignisse, die zur Deletion einzelner DNS-Abschnitte führt. Diese Deletion ist katastrophal und führt zum DiGeorge/velocardiofacial Syndrom. 4
Diese zwei Studien befassten sich mit einzelnen Pseudogenen. Wir müssen mit Vorsicht vorgehen, wenn wir denken, dass wir die Ergebnisse auf alle untranskribierten Pseudogene übertragen können. Nichtsdestotrotz öffnen diese Ergebnisse mindestens die Möglichkeit, dass andere untranskribierten Pseudogensequenzen gleich funktionieren. Wenn die Geschichte uns was bezüglich der Junk DNS zu sagen hat, sind diese zwei Entdeckungen sehr wahrscheinlich Vorboten von dem, was noch kommt. Einfach gesagt, wir entdecken unerwartete Funktion für Pseudogene (und andere Klassen der Junk DNS) immer wieder.
Gemeinsames Design oder gemeinsame Abstammung?
Vor nicht allzu lange wurden geteilte, nicht-funktionsfähige Junk- DNS-Sequenzen in den Genomen der Menschen und Menschenaffen für prima facia Beweise unserer gemeinsamen Entwicklungsgeschichte mit den Menschenaffen gehalten. Die einzige ehrliche Antwort auf die Herausforderung für Schöpfungsmodelle, die Junk-DNS darstellte, war den Gedanken zu äußern, dass wir eines Tages Funktionen für die Junk-DNS-Sequenzen entdecken würden.
Später haben Entdeckungen eine zentrale wissenschaftliche Vorhersage erfüllt, die gleichermaßen von Kreationisten und Verfechter des Intelligent Designs gemacht wurde. Diese ersten Entdeckungen betrafen einzelne, isolierte Pseudogene. Spätere Studien haben gezeigt, dass Pseudogenfunktion überall vorhanden ist. Das hat zu neuen wissenschaftlichen Ideen geführt, z.B. die kompetitive endogene RNA Hypothese, die die Sequenzähnlichkeit der Pseudogene und “intakten” Genen mit Pseudogenfunktion verbindet. Forscher beginnen schon jetzt, funktionelle Rollen für untranskribierte Pseudogene zu identifizieren. Ich sage voraus, dass es nur eine Frage der Zeit ist, bevor Biologen zugeben, dass der Nutzen von untranskribierten Pseudogenen überall vorhanden und tiefgreifend ist.
Die aus dem Schöpfungsmodell entwickelte Interpretation der gemeinsamen Junk DNS-Sequenzen wird stärker und stärker mit jedem Schritt vorwärts. Das gibt mir den Anreiz, diese Frage zu stellen: Wann werden Naturwissenschaftler mit dem Rumscherzen aufhören und dem Ansatz des Schöpfungsmodell einen Sitz am Tisch der Biologie gönnen?
Seth W. Cheetham, Geoffrey J. Faulkner, and Marcel E. Dinger, “Overcoming Challenges and Dogmas to Understand the Functions of Pseudogenes,” Nature Reviews Genetics 21 (17.Dezember.2019): 191–201, doi:10.1038/s41576-019-0196-1.
Cheetham et al., 191–201.
Peng Huang, et al., “Comparative Analysis of Three-Dimensional Chromosomal Architecture Identifies a Novel Fetal Hemoglobin Regulatory Element,” Genes and Development 31, no. 16 (15.August.2017): 1704–13, doi: 10.1101/gad.303461.117.
Laia Vergés et al., “An Exploratory Study of Predisposing Genetic Factors for DiGeorge/Velocardiofacial Syndrome,” Scientific Reports 7 (6.Januar.2017): id. 40031, doi: 10.1038/srep40031.
Stellen Sie Sich vor, Sie reisen durch New England und verfahren Sich. Wenn Sie einen Einheimischen um eine Wegbeschreibung bitten, wird er (oder sie)...
Manchmal kann ich nichts dafür. Ich weiß, es ist der purste Clickbait, aber ich klicke trotzdem. Vor einigen Tagen als Folge einer vorübergehenden Schwäche...